
英文名: Jujuboside A
中文别名:
英文别名:
Cas 号: 55466-04-1
产品编码:BP0813
分子式: C58H94O26
分子量: 1207.36
来源: seeds of Zizyphus jujuba (Chinese date)
纯度: 95%~99%
分析方法: HPLC-DAD or/and HPLC-ELSD
鉴定方法: 质谱(Mass), 核磁(NMR)
包装: 棕色小玻璃瓶,标准包装10mg,20mg,50mg;可以按客户需求包装。
可以满足克级以上大量需求,详情请咨询。
提供相关图谱和分析方法指导,可以提供包括色谱柱,标准品和分析方法在内的含量测定整体服务包,价格优惠。
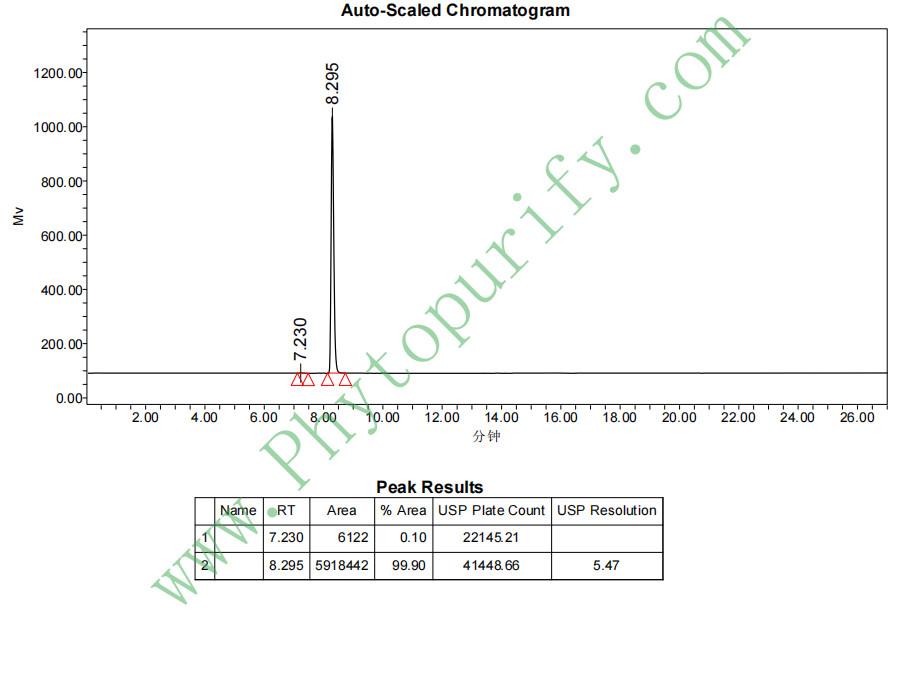
酸枣仁皂苷A的HPLC图谱
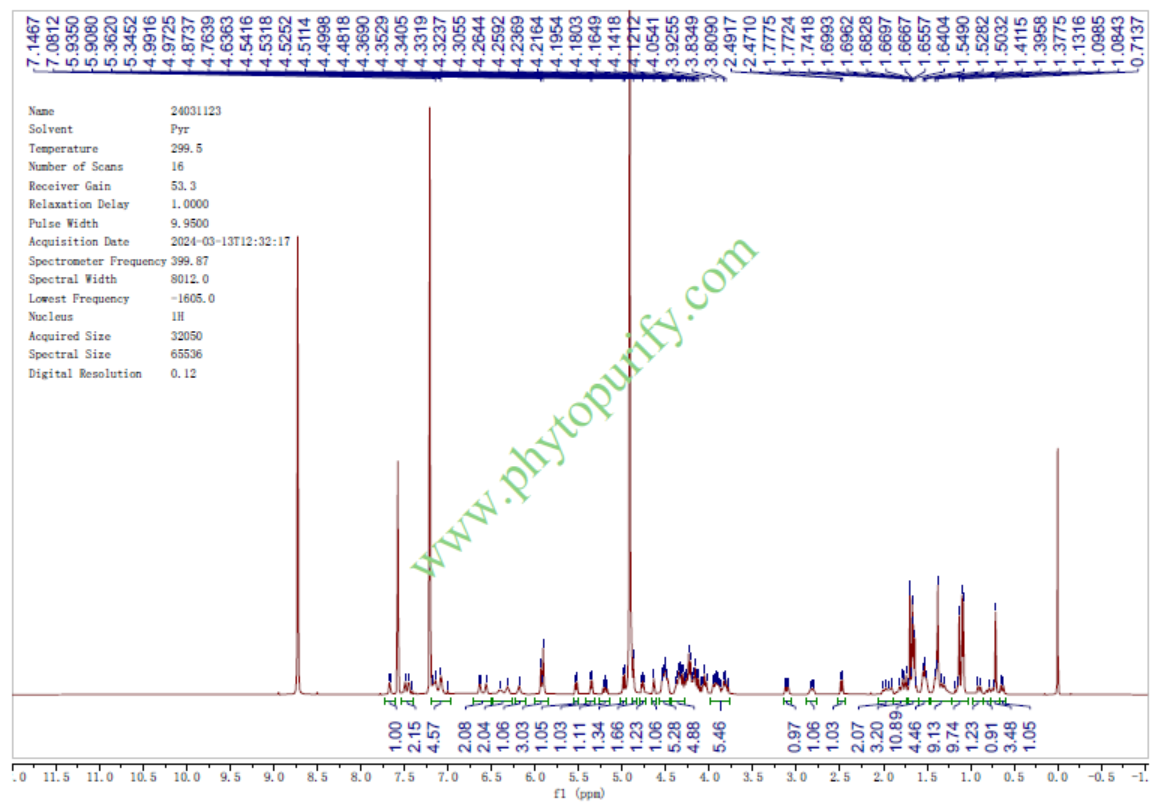
酸枣仁皂苷A的核磁图谱
储存条件:短期保存 2~8℃,长期保存 -20 ~ -80℃
393.98
1.69
1.69
0.19
0.47
0.32
低
66.59
7.74
否
否
否
否
无
无
0.0
是
否
否
否

失眠与焦虑障碍作为现代社会中高发的精神神经系统疾病,严重影响着全球数亿人口的生活质量与身心健康。据世界卫生组织统计,全球约有27%的人口遭受不同程度的睡眠障碍困扰,而焦虑症的终身患病率高达16.6%。在临床治疗中,苯二氮䓬类药物与选择性5-羟色胺再摄取抑制剂虽为一线用药,但其伴随的依赖性、耐受性、认知功能损害及戒断反应等副作用,促使研究者不断寻求更安全、更有效的替代疗法。在此背景下,源于传统中医药的天然产物因其多靶点、低毒性的特点,日益成为神经精神药物研发的重要源泉。
酸枣仁(Ziziphus jujuba Mill. var. spinosa (Bunge) Hu ex H.F.Chow)作为中医临床使用历史最为悠久的安神药物之一,始载于《神农本草经》,被列为上品,具有养心补肝、宁心安神、敛汗生津的功效,主治虚烦不眠、惊悸多梦、体虚多汗等症。现代药理学研究证实,酸枣仁的主要活性成分包括皂苷类、黄酮类、生物碱类及脂肪酸类化合物,其中酸枣仁皂苷A(Jujuboside A,JuA)被认为是其镇静催眠与抗焦虑作用的核心物质基础。
酸枣仁皂苷A是一种达玛烷型三萜皂苷,化学结构复杂,分子量高达1207.36 Da,具有独特的糖链修饰模式。自20世纪80年代首次从酸枣仁中分离鉴定以来,JuA一直是天然产物化学与神经药理学领域的研究热点。近年来,随着分子生物学与系统药理学技术的发展,JuA在调控神经递质系统、调节突触可塑性、影响神经炎症反应等方面的作用机制逐步被揭示,其在失眠、焦虑、认知障碍等多种神经系统疾病中的治疗潜力也得到广泛关注。本文将从化学结构、植物来源、药理活性、分子机制、成药性评价及临床应用前景等方面,对酸枣仁皂苷A的研究进展进行系统综述,以期为该天然产物的深入开发与临床转化提供参考。
酸枣仁皂苷A的化学名为(3β,16β,20R)-16,23:16,30-二环氧-20-羟基达玛烷-24-烯-3-基-O-β-D-吡喃木糖基-(1→2)-O-β-D-吡喃葡萄糖基-(1→3)-O-α-L-吡喃鼠李糖基-(1→2)-β-D-吡喃葡萄糖苷,CAS登记号为55466-04-1。其分子式为C₅₈H₉₄O₂₆,分子量为1207.3640 Da,属于典型的高分子量天然糖苷类化合物。
从结构特征来看,JuA的苷元为达玛烷型四环三萜骨架,C-3位羟基通过糖苷键连接一条由四个单糖组成的线性糖链,其连接顺序为:葡萄糖(Glc)-鼠李糖(Rha)-葡萄糖(Glc)-木糖(Xyl),其中木糖以β-(1→2)键连接于末端葡萄糖,鼠李糖以α-(1→3)键连接于中间葡萄糖。此外,苷元部分在C-16和C-23位之间形成一个环氧桥,C-16和C-30位之间形成另一个环氧桥,这种独特的环醚结构赋予了JuA特殊的空间构象与生物活性。C-20位为叔羟基,C-24-25位为双键,这些官能团可能参与分子与靶蛋白的相互作用。
在理化性质方面,JuA的脂水分配系数(LogP)为1.6871,表明其具有一定的亲脂性但总体偏向亲水。拓扑极性表面积(TPSA)高达393.98 Ų,远高于口服药物通常推荐的140 Ų上限,这主要归因于分子中大量的羟基与糖苷键氧原子。水溶性参数为0.1860 mg/mL,属于微溶范畴。值得注意的是,JuA的血脑屏障穿透能力被评估为“低”,这与其高分子量、高极性表面积及亲水性特征相符。然而,后续研究证实JuA仍可在中枢神经系统中发挥药理作用,提示可能存在主动转运机制或通过代谢产物发挥作用。此外,hERG抑制试验结果为阴性,表明JuA不具有心脏毒性风险;Ames试验结果为0.0,提示其无致突变性,初步安全性良好。
酸枣仁皂苷A主要来源于鼠李科枣属植物酸枣(Ziziphus jujuba Mill. var. spinosa)的干燥成熟种子。酸枣广泛分布于中国华北、西北及东北地区,以河北、山西、陕西、山东等省份产量最大。此外,同属植物滇刺枣(Ziziphus mauritiana Lam.)及大枣(Ziziphus jujuba Mill.)的种子中也含有少量JuA,但含量远低于酸枣仁。酸枣仁中JuA的含量受品种、产地、采收时间及加工方式等因素影响,通常在0.03%~0.15%之间(以干燥品计),属于含量较低的活性成分。
传统的酸枣仁皂苷提取多采用乙醇回流法。具体工艺为:将干燥酸枣仁粉碎后,用60%~80%乙醇在60~80℃条件下回流提取2~3次,每次1~2小时,合并提取液,减压浓缩后得到浸膏。该浸膏经石油醚脱脂、正丁醇萃取,可富集皂苷类成分。进一步纯化可采用大孔吸附树脂柱层析(如D101、AB-8型树脂),以水-乙醇梯度洗脱,收集30%~70%乙醇洗脱部位,经浓缩干燥得到总皂苷粗品。JuA的分离纯化通常需要结合硅胶柱层析、ODS反相柱层析及制备型高效液相色谱(HPLC)等技术。例如,以氯仿-甲醇-水(65:35:10,下层)为流动相进行硅胶柱层析,再以乙腈-水(30:70)为流动相进行制备型HPLC分离,可获得纯度达98%以上的JuA单体。
近年来,为提高提取效率与产物纯度,多种现代提取技术被应用于JuA的制备。超声辅助提取利用空化效应破坏细胞壁结构,可在30~60分钟内完成提取,JuA提取率较传统回流法提高20%~30%。微波辅助提取通过极性分子在微波场中的快速振动产生内热,使提取时间缩短至10~20分钟,且溶剂用量减少。超临界流体萃取(以CO₂为溶剂,添加乙醇为夹带剂)则具有绿色环保、选择性高的优点,但设备成本较高。此外,酶辅助提取(使用纤维素酶、果胶酶预处理原料)可破坏细胞壁多糖骨架,使JuA更易释放,提取率可提升40%以上。这些新技术的应用为JuA的高效制备与产业化提供了技术支撑。
JuA的镇静催眠活性是其最为经典且研究最为深入的功效。早在20世纪90年代,日本学者就发现腹腔注射JuA可显著延长戊巴比妥钠诱导的小鼠睡眠时间,并减少自主活动次数。后续研究在多种动物模型中验证了这一作用:口服或腹腔给予JuA(5~40 mg/kg)可剂量依赖性地缩短睡眠潜伏期、延长睡眠持续时间,且不引起肌肉松弛和共济失调等苯二氮䓬类药物的典型副作用。值得注意的是,JuA的催眠作用呈现独特的“双向调节”特征——在正常生理状态下对睡眠结构影响较小,但在应激或病理状态下(如慢性束缚应激、对氯苯丙氨酸诱导的失眠模型)可显著改善睡眠质量,提示其可能通过调节内源性睡眠-觉醒稳态发挥作用。
脑电图分析显示,JuA可增加非快速眼动睡眠(NREM)时间,尤其是慢波睡眠(SWS)的持续时间,而对快速眼动睡眠(REM)影响较小。这种作用模式与经典的GABA能药物不同,后者往往同时抑制REM睡眠。此外,JuA可降低脑电图δ波功率密度,提示其可能通过调节丘脑-皮层环路的功能活动来促进睡眠。
JuA的抗焦虑活性在多种行为学模型中得到证实。在高架十字迷宫实验中,JuA(10~20 mg/kg)可显著增加小鼠进入开放臂的次数和时间比例,表明其具有抗焦虑效应。在明暗箱实验、 Vogel冲突实验及社会互作实验中,JuA均表现出剂量依赖性的抗焦虑作用,且效果与阳性对照药地西泮相当,但无明显的镇静或运动抑制副作用。值得注意的是,JuA的抗焦虑作用在慢性应激模型中尤为突出:长期给予JuA可逆转慢性不可预测温和应激(CUMS)诱导的大鼠焦虑样行为,同时改善下丘脑-垂体-肾上腺(HPA)轴功能紊乱。
除镇静催眠与抗焦虑作用外,JuA的神经保护活性近年来受到广泛关注。在谷氨酸诱导的兴奋性毒性模型中,JuA可显著降低原代皮层神经元的凋亡率,抑制线粒体膜电位下降和细胞色素c释放。在β-淀粉样蛋白(Aβ)诱导的阿尔茨海默病细胞模型中,JuA可减少Aβ聚集、降低Tau蛋白过度磷酸化水平,并改善突触蛋白的表达。此外,JuA对脑缺血再灌注损伤、帕金森病模型及癫痫模型均表现出保护作用,其机制涉及抗氧化应激、抗神经炎症及抗凋亡等多条通路。
近年研究还发现JuA具有调节肠道菌群、改善代谢紊乱的作用。在抗生素诱导的肠道菌群失调小鼠中,JuA可恢复乳酸杆菌和双歧杆菌的丰度,降低条件致病菌的比例,同时改善肠屏障功能。此外,JuA对心肌缺血再灌注损伤、肝纤维化及肾损伤也表现出一定的保护作用,提示其可能具有多系统、多靶点的药理活性谱。
GABA(γ-氨基丁酸)是中枢神经系统中最主要的抑制性神经递质,其受体GABAA是苯二氮䓬类药物和多种镇静催眠药物的作用靶点。JuA对GABA能系统的调控是其镇静催眠作用的核心机制之一。研究表明,JuA可增强GABA诱导的氯离子内流,增加GABAA受体的开放频率,但作用方式与苯二氮䓬类药物不同:JuA不与苯二氮䓬结合位点竞争,而是通过变构调节增强GABA与受体的亲和力。分子对接与突变分析显示,JuA可能结合于GABAA受体的α1/β2亚基界面,与β2亚基的Tyr157、Thr202及α1亚基的Phe64等氨基酸残基形成氢键和疏水相互作用。
在受体亚基组成方面,JuA对含有α1、β2、γ2亚基的GABAA受体(GABRA1、GABRB2、GABRG2)表现出较高的选择性。这一特征具有重要的药理学意义:α1亚基介导镇静催眠作用,而α2/α3亚基主要参与抗焦虑和肌肉松弛作用。JuA对α1亚基的选择性可能解释其为何在产生催眠作用的同时不引起明显的肌肉松弛和共济失调。此外,JuA还可上调GABAA受体各亚基的蛋白表达水平,长期给药可增加皮层和海马中GABRA1、GABRB2、GABRG2的表达,这可能是其产生持续抗焦虑作用的分子基础。
5-羟色胺(5-HT)系统在情绪调节、睡眠-觉醒周期及焦虑反应中发挥关键作用。JuA对5-HT系统的调控涉及多个靶点。首先,JuA可抑制5-羟色胺转运体(SLC6A4,即SERT)的活性,减少突触间隙中5-HT的再摄取,从而增加突触间隙5-HT浓度。这种作用与选择性5-羟色胺再摄取抑制剂(SSRIs)类似,但强度较弱,可能避免了SSRIs常见的起效延迟和性功能障碍等副作用。
其次,JuA可调节5-HT受体的表达与功能。研究发现,JuA可上调5-HT1A受体(HTR1A)的表达,同时下调5-HT2A受体(HTR2A)的表达。5-HT1A受体是突触前和突触后抑制性受体,其激活可降低5-HT能神经元的放电频率,产生抗焦虑和抗抑郁效应;而5-HT2A受体激活则与焦虑和失眠相关。JuA对这两种受体的差异性调节,可能协同产生抗焦虑和镇静催眠作用。此外,JuA还可增强5-HT1A受体与G蛋白的偶联效率,提高下游信号通路的转导活性。
下丘脑-垂体-肾上腺(HPA)轴是机体应激反应的核心调节系统,其功能紊乱与失眠和焦虑密切相关。JuA可显著降低慢性应激模型大鼠血清中促肾上腺皮质激素释放激素(CRH)、促肾上腺皮质激素(ACTH)和皮质酮的水平,抑制HPA轴的过度激活。机制研究表明,JuA可上调海马中糖皮质激素受体(GR)的表达,增强GR对HPA轴的负反馈调节;同时抑制下丘脑室旁核中CRH神经元的激活,减少CRH的合成与释放。这种对HPA轴的调节作用可能是JuA改善应激相关睡眠障碍和焦虑症状的重要机制。
神经炎症和氧化应激是失眠与焦虑的重要病理机制。JuA可抑制脂多糖(LPS)或Aβ诱导的小胶质细胞激活,减少肿瘤坏死因子-α(TNF-α)、白细胞介素-1β(IL-1β)和白细胞介素-6(IL-6)等促炎因子的释放,同时增加抗炎因子IL-10的表达。其抗炎机制涉及抑制Toll样受体4(TLR4)/核因子-κB(NF-κB)信号通路,以及激活核因子E2相关因子2(Nrf2)/抗氧化反应元件(ARE)通路。在氧化应激方面,JuA可降低活性氧(ROS)和丙二醛(MDA)水平,提高超氧化物歧化酶(SOD)、谷胱甘肽过氧化物酶(GSH-Px)和过氧化氢酶(CAT)的活性,从而保护神经元免受氧化损伤。
慢性失眠和焦虑常伴随海马和前额叶皮层突触可塑性的损害。JuA可逆转应激诱导的海马CA1区树突棘密度降低和突触蛋白(如PSD-95、Synapsin I)表达下降,增强长时程增强(LTP)效应。机制研究表明,JuA可通过激活脑源性神经营养因子(BDNF)/酪氨酸激酶受体B(TrkB)/cAMP反应元件结合蛋白(CREB)信号通路,促进突触发生和神经发生。此外,JuA还可调节谷氨酸能突触传递,降低NMDA受体介导的兴奋性毒性,维持兴奋/抑制平衡。
JuA的药代动力学研究主要基于大鼠和小鼠模型。口服给药后,JuA的吸收较差,绝对生物利用度约为1%~3%,这与其高分子量、高极性及低渗透性密切相关。血浆药物浓度-时间曲线呈双峰现象,提示可能存在肠肝循环或肠道菌群代谢。达峰时间(Tmax)约为1~2小时,消除半衰期(t₁/₂)约为3~5小时。静脉给药后,JuA在体内分布广泛,以肝脏、肾脏和肺脏中浓度最高,脑组织中浓度较低,但可通过血脑屏障,脑/血浆浓度比约为0.05~0.1。
代谢方面,JuA在体内主要经历去糖基化代谢。肠道菌群可水解JuA的糖链,生成次级苷元(如酸枣仁皂苷B、酸枣仁皂苷元)及单糖。这些代谢产物可能具有不同的药理活性,部分代谢物(如酸枣仁皂苷元)已被证实具有更强的镇静活性。肝脏中的细胞色素P450酶(CYP3A4为主要亚型)也参与JuA的氧化代谢。排泄途径以胆汁和粪便为主,尿液中仅检测到少量原型药物。
从成药性角度看,JuA面临的主要挑战是口服生物利用度低和血脑屏障穿透能力差。其分子量(1207 Da)远超口服药物的“五规则”(MW<500),TPSA(394 Ų)也远高于口服药物通常推荐的140 Ų上限,这些参数预示其口服吸收和中枢分布存在障碍。然而,JuA在体内仍能发挥显著的中枢药理作用,提示可能存在以下机制:(1)肠道菌群代谢产生的活性代谢物可穿透血脑屏障;(2)通过载体介导的转运(如有机阴离子转运多肽OATP)实现肠道吸收和脑部分布;(3)作用于外周靶点(如肠道神经系统、迷走神经)间接调节中枢功能。
在安全性方面,JuA表现出良好的耐受性。急性毒性试验显示,小鼠口服JuA的LD₅₀大于5000 mg/kg,腹腔注射的LD₅₀约为800 mg/kg。亚慢性毒性试验(28天重复给药)未观察到明显的器官毒性或血液学异常。hERG抑制试验阴性,提示无心脏毒性风险;Ames试验阴性,表明无遗传毒性。这些数据支持JuA作为先导化合物进行结构优化和制剂开发的潜力。
为提高JuA的成药性,研究者开展了多种结构修饰和制剂研究。前药策略是改善口服吸收的有效手段:通过在JuA的羟基上引入酯基或磷酸基团,可提高脂溶性,促进肠道吸收,并在体内经酶解释放原型药物。例如,JuA的琥珀酸酯前药在大鼠中的口服生物利用度提高了5倍以上。纳米制剂技术也显示出良好前景:聚乳酸-羟基乙酸共聚物(PLGA)纳米粒可包封JuA,提高其稳定性,延长循环时间,并增强脑部递送。脂质体、固体脂质纳米粒及磷脂复合物等制剂形式也在研究中,初步结果显示可显著提高JuA的口服吸收和脑靶向性。
基于JuA的镇静催眠作用及其独特的药理学特征(不引起肌肉松弛、无依赖性、双向调节睡眠结构),其在失眠症治疗中具有广阔的应用前景。目前,以酸枣仁总皂苷为主要成分的中成药(如酸枣仁汤、酸枣仁胶囊)已在临床广泛使用,但JuA单体的制剂尚未上市。未来,JuA有望开发为治疗慢性失眠、尤其是伴有焦虑症状的失眠症的新型药物。其“应激选择性”的催眠特点,使其特别适合用于压力相关失眠和轮班工作导致的睡眠节律紊乱。
JuA的抗焦虑作用与SSRIs相当,但起效更快(数小时至数天 vs. 数周),且无性功能障碍、体重增加等常见副作用。对于广泛性焦虑症、社交焦虑症及惊恐障碍,JuA可能提供一种新的治疗选择。此外,JuA对HPA轴的调节作用使其在应激相关障碍(如创伤后应激障碍PTSD)中具有潜在应用价值。
JuA的神经保护、抗炎和抗氧化作用提示其在阿尔茨海默病、帕金森病等神经退行性疾病中的潜在应用。特别是,失眠和焦虑是这些疾病常见的非运动症状,JuA可能同时改善认知功能和情绪睡眠障碍,实现“一药多效”。然而,目前研究主要基于细胞和动物模型,临床证据尚需积累。
尽管JuA的研究取得了显著进展,但其临床转化仍面临多重挑战。首先,口服生物利用度低是最大的瓶颈,需要开发高效的前药或纳米制剂系统。其次,JuA的多靶点作用机制虽然有利于发挥综合疗效,但也增加了副作用预测和药物相互作用评估的复杂性。第三,目前缺乏高质量的临床随机对照试验,其临床疗效和安全性数据尚不充分。未来研究应重点关注:(1)开发高生物利用度的JuA衍生物或制剂;(2)利用系统药理学和网络药理学方法,阐明JuA的“多成分-多靶点-多通路”作用网络;(3)开展设计严谨的临床研究,验证其治疗失眠和焦虑的有效性和安全性;(4)探索JuA与其他天然产物或化学药物的协同作用,开发复方制剂。
酸枣仁皂苷A作为传统安神中药酸枣仁的核心活性成分,历经数十年的研究,其化学结构、理化性质、药理活性及分子机制已得到较为系统的阐明。从GABA能系统、5-HT能系统到HPA轴、神经炎症和突触可塑性,JuA通过多靶点、多通路的协同调控,发挥镇静催眠、抗焦虑和神经保护作用。其独特的药理学特征——无肌肉松弛副作用、不产生依赖性、双向调节睡眠结构——使其区别于传统的苯二氮䓬类药物,具有成为新型镇静催眠和抗焦虑药物的潜力。
然而,JuA的高分子量、高极性导致的低口服生物利用度和低血脑屏障穿透能力,是其临床转化的主要障碍。未来,通过前药设计、纳米制剂技术及结构优化,有望突破这些成药性瓶颈。同时,随着对JuA代谢产物、肠道菌群相互作用及外周-中枢通讯机制的深入研究,我们对其体内作用模式的理解将更加全面。从“酸枣仁安神”的传统经验到JuA的现代药理学诠释,这一天然产物不仅为失眠和焦虑的治疗提供了新的候选分子,也展示了传统中医药智慧与现代药物科学融合的典范。我们有理由相信,在不久的将来,酸枣仁皂苷A或其衍生物将进入临床,为全球数以亿计的失眠和焦虑患者带来新的治疗希望。

版权所有:© 成都普瑞法科技开发有限公司(2015)备案号:蜀ICP备15035167号-1 客服热线:400-829-7929
技术支持:南京库价















